
La Agencia Europea de Medicamentos (EMA) anunció en octubre de 2024 la adopción de la tan esperada directriz sobre la Calidad y Equivalencia de Productos Tópicos. El documento final se redactó en 2018 y entrará en vigor en abril de 2025 con el título oficial de « Directriz sobre la calidad y equivalencia de productos cutáneos de aplicación y acción local ».
Nuestros departamentos de TRIV y PTIV y de Garantía de Calidad han resumido los aspectos más relevantes de la guía de la EMA en relación con la TRIV y la PTIV, dividiendo los mismos entre las recomendaciones de calidad y la sección de equivalencia terapéutica. Finalmente, han compartido la experiencia del Grupo Kymos trabajando en cumplimiento con la misma desde su último borrador en 2018.
¿Cuál es la nueva directriz de la EMA sobre productos tópicos para 2024?
Como sugiere el título, esta directriz se refiere a medicamentos de aplicación y acción local para uso cutáneo, pero también puede ser relevante para otros productos, como preparaciones para uso auricular u ocular, y productos vaginales de acción local. Este nuevo reglamento se divide en recomendaciones de calidad y equivalencia e introduce un enfoque más estructurado para la evaluación de productos cutáneos.
La segunda parte relativa a la equivalencia no se aplica a medicamentos biológicos, medicamentos a base de plantas, productos cuya equivalencia en cuanto a eficacia se haya demostrado mediante ensayos clínicos, ni a productos cuya forma farmacéutica de prueba y de referencia no sea la misma. Esta exclusión es especialmente importante cuando se trata de formas farmacéuticas de absorción sistémica, como los parches transdérmicos.
Con estas recientes actualizaciones, la EMA enfatiza un enfoque más estructurado y gradual que debería simplificar el proceso de demostrar la equivalencia terapéutica centrándose en métodos in vitro (prueba de liberación in vitro o IVRT y prueba de permeación in vitro o IVPT) y farmacocinéticos (PK) que presentan alternativas que ahorran costos y tiempo a los estudios clínicos.
Recomendaciones de calidad para productos tópicos
Las recomendaciones de calidad de la guía se aplican a las nuevas solicitudes de autorización de comercialización y a los cambios posteriores a la aprobación de productos no contemplados en otras directrices ni en las normas pertinentes de la Farmacopea. En cuanto a la TRIV y la TPIV, los aspectos más relevantes de la guía son:
Desarrollo farmacéutico
-
Desarrollo de formulaciones
El desarrollo de la formulación debe estar alineado con el QTTP (Perfil de Producto Objetivo de Calidad), con pruebas adecuadas para caracterizar y controlar los CQA (Atributos Críticos de Calidad) como la facilidad de administración, la duración del uso y el desempeño del producto como la disolución, la IVRT y, si corresponde, la IVPT.
-
Caracterización del producto
La caracterización es necesaria para facilitar la gestión del ciclo de vida y, de ser necesario, la equivalencia del producto. Las pruebas clave de rendimiento deben incluir la disolución de suspensiones, la IVRT y la IVPT, si es necesario. Se debe demostrar que el rendimiento del producto es estable durante el almacenamiento.
Estrategia de control
Los controles de calidad de la calidad (CQA), cruciales para el control de la liberación del fármaco, deben gestionarse cuidadosamente con pruebas como la IVRT y la IVPT (si procede). Se pueden utilizar otros parámetros (p. ej., microscopía, DSC, reología) si se demuestra que son más discriminantes para controlar la liberación del fármaco. Además, cualquier límite de rendimiento de las pruebas (p. ej., disolución, IVRT) incluido en la especificación debe justificarse con referencia a lotes clínicos con eficacia y seguridad comprobadas.
Programa de Estabilidad
Las pruebas de estabilidad deben garantizar la calidad y la eficacia del producto a lo largo del tiempo, y la IVRT u otras pruebas de rendimiento deben confirmar la consistencia de la vida útil.
Recomendaciones de equivalencia para productos tópicos
Esta parte de la guía se aplica a nuevos productos cutáneos que buscan demostrar equivalencia terapéutica con un medicamento existente. También es aplicable a cambios posteriores a la aprobación cuando se prevé un posible impacto en la calidad, la seguridad o la eficacia tras una evaluación de riesgos.
La guía también establece específicamente que “en el caso de solicitudes que se basan en literatura para demostrar la seguridad y eficacia del medicamento, la relevancia de la literatura debe respaldarse con datos de equivalencia con el producto descrito en la literatura”.
Como se mencionó anteriormente, la EMA recomienda un enfoque gradual para demostrar la equivalencia. Esto permite a los fabricantes farmacéuticos saber de antemano qué pruebas realizar para sus productos (formulaciones simples, como soluciones o geles, o formulaciones complejas, como emulsiones), y su objetivo principal es omitir los estudios clínicos completos.
Al decidir qué enfoque y árbol de decisión seleccionar, la EMA tiene en cuenta la composición cualitativa, la composición cuantitativa y la caracterización fisicoquímica y estructural de los productos cutáneos:
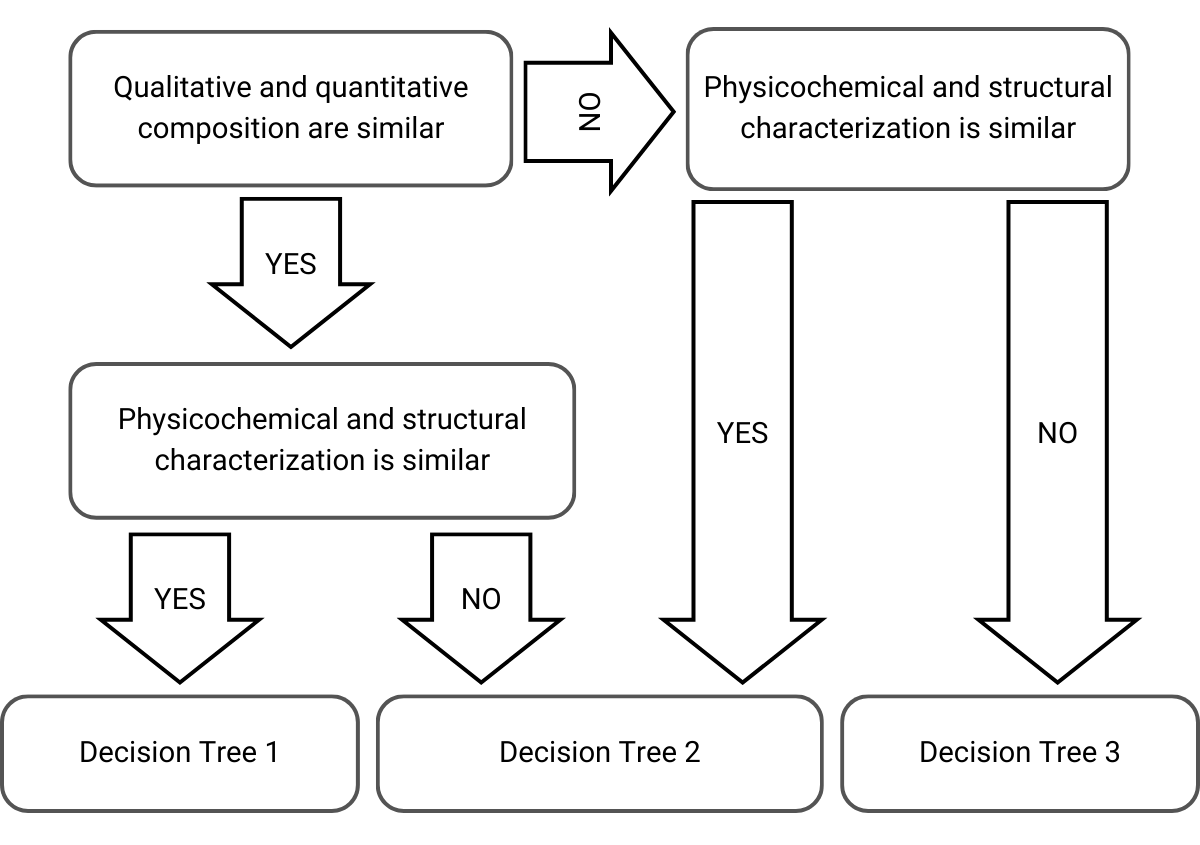
Figura 1) Selección del árbol de decisiones en el enfoque paso a paso adaptado de la directriz de la EMA
Luego, dependiendo de estas consideraciones, el enfoque paso a paso es el siguiente en los siguientes árboles de decisión que conducen a puentes aceptables o rechazos:
Árbol de decisión 1: Misma composición cualitativa y cuantitativa, y misma caracterización fisicoquímica y estructural
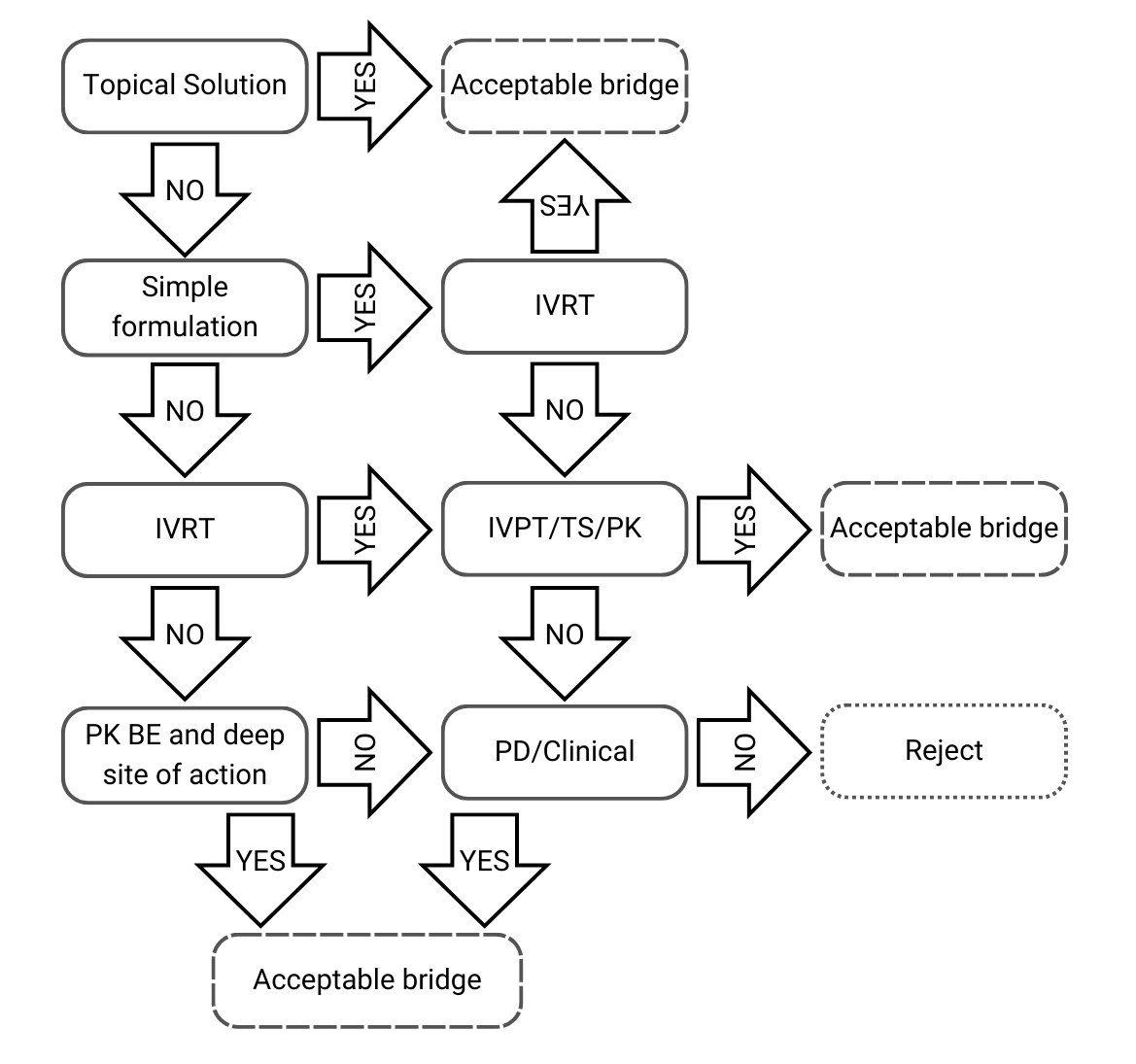
Figura 2) Árbol de decisión 1 de la directriz de la EMA
Árbol de decisión 2: Pequeñas diferencias en las composiciones cualitativas y cuantitativas, y/o caracterización fisicoquímica y estructural
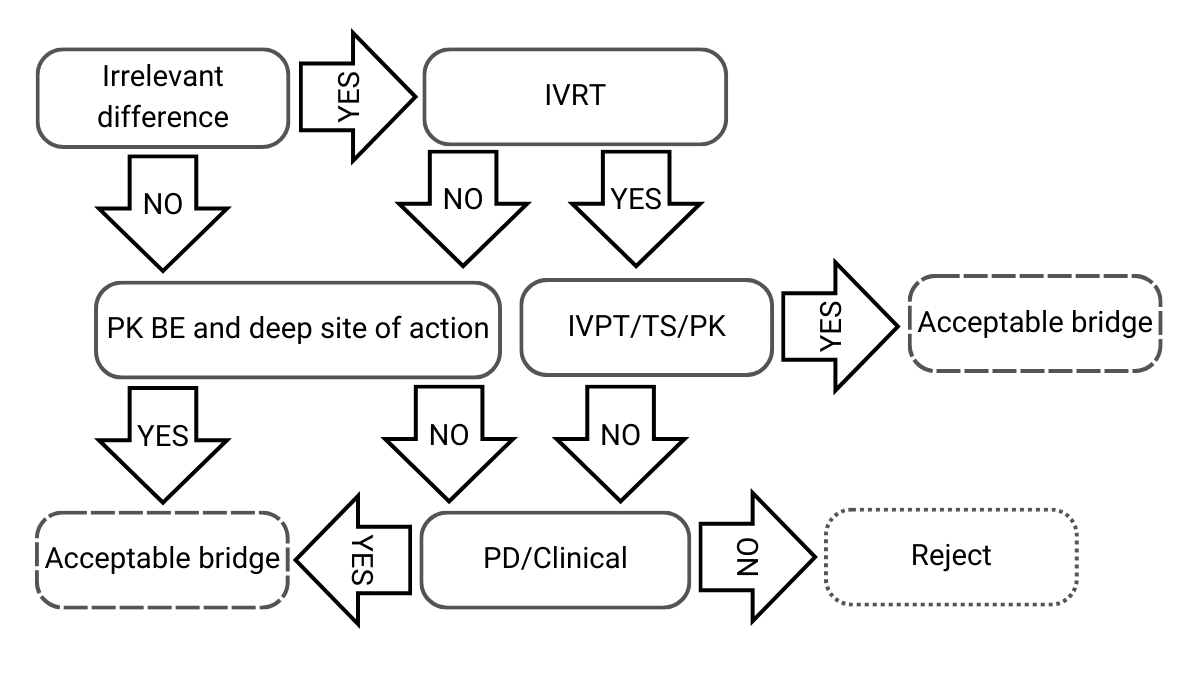
Figura 3) Árbol de decisión 2 de la directriz de la EMA
Árbol de decisión 3: Diferentes composiciones cualitativas y cuantitativas y diferente caracterización fisicoquímica y estructural
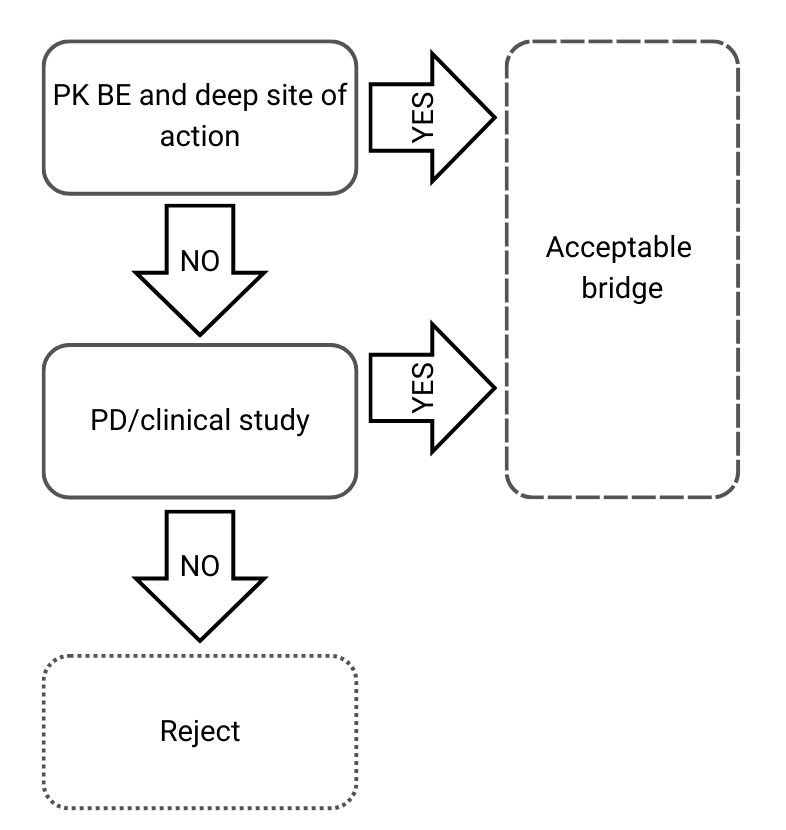
Figura 4) Árbol de decisión 3 de la directriz de la EMA
Como se ve en estos árboles de decisión, dependiendo de las similitudes entre los productos, la EMA ofrece diferentes enfoques graduales que los fabricantes deberían considerar.
Experiencia del Grupo Kymos con estudios IVRT e IVPT
El Grupo Kymos cuenta con un equipo especializado que trabaja en la liberación de fármacos y técnicas para productos tópicos desde 2017 y cuenta con una amplia experiencia en estudios de IVRT e IVPT. Nuestros métodos se han desarrollado de conformidad con la versión preliminar de esta misma guía de 2018, y nuestros científicos ya están familiarizados con esta versión final.
Hemos dividido nuestro catálogo de servicios de liberación de fármacos y absorción percutánea en dos grupos principales:
- IVRT: Medimos la cantidad y la velocidad de liberación de fármacos mediante membranas artificiales para desarrollar y validar métodos para diferentes formulaciones. Estos métodos pueden emplearse en estudios comparativos para evaluar la equivalencia y también para el control de calidad de los lotes de fabricación.
- IVPT: Medimos las cantidades permeadas transdérmicamente, las tasas de flujo y la distribución de capas con muestras de piel para estudios de bioequivalencia. También podemos contribuir a la optimización y comparación de formulaciones mediante estudios de absorción percutánea.
Nuestro laboratorio es una de las pocas instalaciones europeas que ofrecen ensayos de absorción percutánea certificados tanto por GLP como por GMP para productos cutáneos con los últimos instrumentos automatizados de difusión vertical Hanson Phoenix (prueba de células de Franz).
Con un profundo conocimiento de las nuevas directrices, nuestro equipo científico está preparado para apoyar a los clientes desde el desarrollo de la formulación hasta la presentación reglamentaria de nuevas solicitudes de comercialización, pero también para productos cutáneos que quieran demostrar equivalencia con productos medicinales existentes siguiendo el enfoque gradual de la EMA.
Conclusiones
La adopción de la nueva directriz de la EMA sobre la calidad y equivalencia de productos tópicos supone un gran avance hacia un enfoque más armonizado, pero también más estructurado, para el análisis de este tipo de medicamentos. Sus recomendaciones graduales para demostrar la equivalencia mediante técnicas como la TRIV y la TPIV agilizan el proceso de comercialización de nuevos genéricos sin necesidad de estudios clínicos costosos y prolongados.
Como CRO líder en IVRT e IVPT, Kymos Group y su equipo están listos para apoyar a los clientes y socios en cada paso, asegurando un camino sin problemas hacia la aprobación del mercado en Europa bajo las últimas pautas.
Si necesita más información sobre las directrices de la EMA o asistencia con sus proyectos de absorción percutánea, póngase en contacto con commercial@kymos.com . Con gusto le brindaremos asesoramiento y apoyo detallados.
